

Living with VLCAD
Living with Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency can feel overwhelming, especially after a new diagnosis. This page is designed to provide trusted, easy-to-understand information, resources, and guidance for individuals, caregivers, and families navigating life with VLCAD and related long-chain fatty acid oxidation disorders (LC-FAOD).

What is VLCAD Deficiency?
Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) deficiency is a rare genetic metabolic disorder that affects how the body produces energy. It belongs to a group of conditions called long-chain fatty acid oxidation disorders (LC-FAOD), which prevent the body from properly breaking down certain fats for fuel.
The body normally uses both sugar (glucose) and fat for energy. During times when glucose is limited, such as sleeping overnight, fasting, illness, or exercise, the body switches to burning stored fat to keep energy levels stable.
For people with VLCAD deficiency, this process does not work properly. The body cannot efficiently break down long-chain fatty acids, which can lead to dangerous energy shortages when the body needs extra fuel.
These energy shortages can affect organs that require large amounts of energy, including the heart, muscles, and liver.
Understanding Long-Chain Fatty Acid Oxidation Disorders (LC-FAOD)
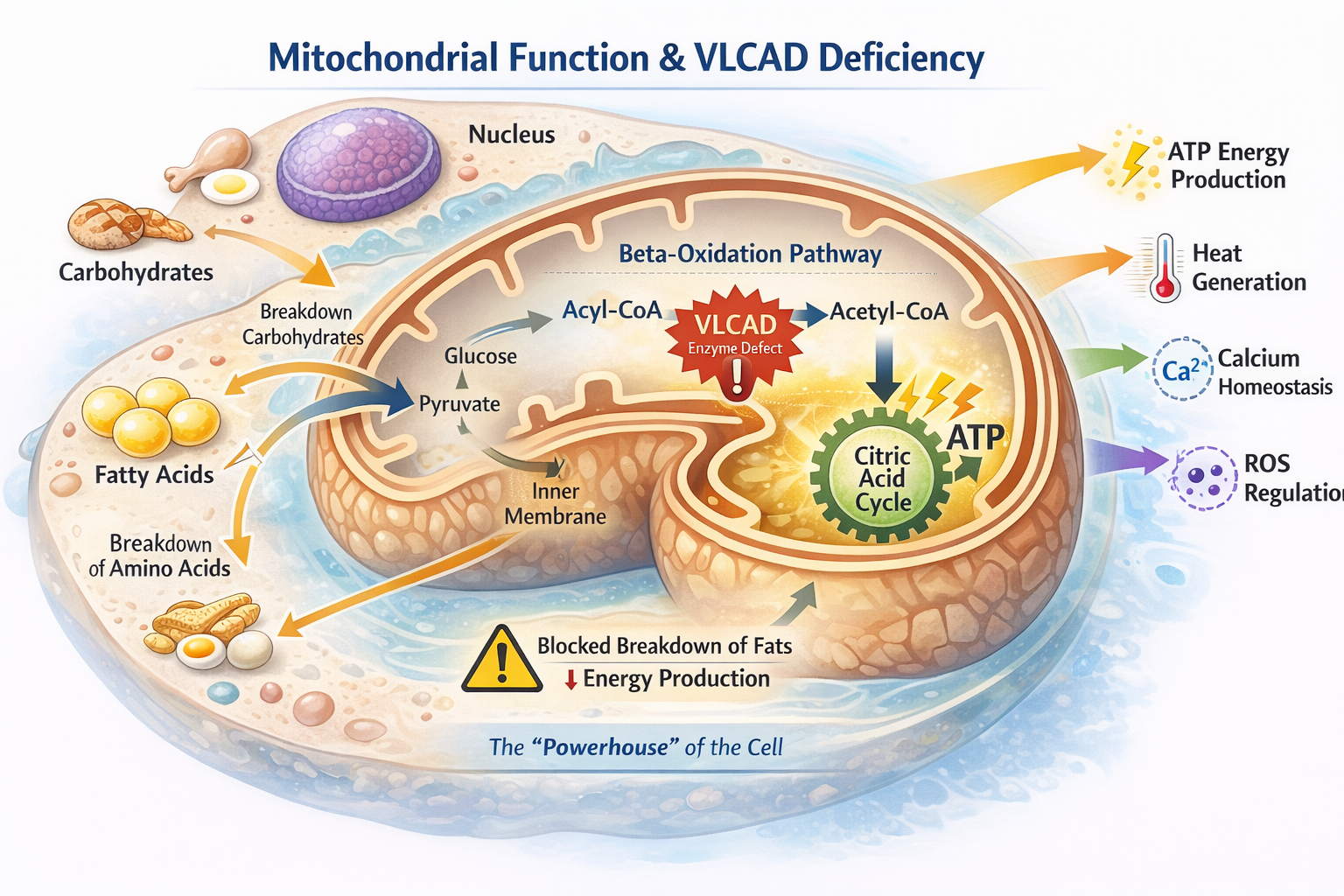
Long-chain fatty acid oxidation disorders (LC-FAOD) are a group of rare inherited metabolic conditions that affect how the body converts fat into energy. This process normally occurs inside the mitochondria, often referred to as the “power plants” of the cell. When the body runs low on glucose, mitochondria help break down stored fat to create energy. In individuals with LC-FAOD, one of the enzymes needed for this process does not function correctly. As a result, the body cannot properly use certain fats for energy. Energy shortages are most likely to occur during:
Fasting or long periods without food
Illness or infection
Intense physical activity
Periods of rapid growth
Physical or metabolic stress
Because the heart, muscles, and liver rely heavily on energy, these organs are often the most affected.
The mitochondria are tiny structures inside our cells that act like the body’s energy factories. Their main job is to take nutrients from food, such as carbohydrates and fats, and convert them into usable energy that powers the heart, muscles, brain, and organs.
When a person eats, their body stores energy from food. Between meals, during sleep, illness, exercise, or growth, the body switches to using fat as a major fuel source. This fat must be processed inside the mitochondria to create steady energy.
To do this, the mitochondria use a series of special helper proteins called enzymes that break down fats step-by-step to release energy for the body. One of these important enzymes, very-long-chain acyl-CoA dehydrogenase (VLCAD), helps break down certain fats so the body can keep making energy when it isn’t eating.
How the Mitochondria Works?
How does VLCAD affect Mitochondria?
In VLCAD deficiency, the mitochondria cannot properly break down long-chain fats because a specific enzyme called VLCAD does not function as it should. This creates a block in the body’s energy pathway, making it difficult to efficiently access stored fat for fuel. As a result, the body can run out of energy more quickly during illness, fasting, stress, or increased physical activity.
When the mitochondria are unable to produce enough energy, it can lead to complications such as low blood sugar (hypoglycemia), fatigue and low energy tolerance, muscle breakdown (rhabdomyolysis), and in some individuals, heart-related complications. This is why consistent nutrition, prescribed medical therapies, and specialized dietary management are essential to help provide a steady and reliable energy source for individuals living with VLCAD.

VLCAD is the most common long-chain fatty acid oxidation disorder. It is estimated to occur in approximately 1 in 30,000 to 100,000 newborns worldwide. Because VLCAD is included in many newborn screening programs, more individuals are being identified early, allowing treatment and management to begin before serious symptoms develop.
Causes and Genetics of VLCAD Deficiency
VLCAD deficiency is caused by changes (mutations) in a gene called ACADVL.
Genes act as instructions that tell the body how to produce proteins and enzymes needed for normal cell function. The ACADVL gene provides instructions for making the VLCAD enzyme, which is responsible for breaking down very long-chain fatty acids inside the mitochondria, the structures in cells that generate energy. This process is part of fatty acid oxidation, a metabolic pathway the body uses to convert stored fat into energy when glucose levels are low. This typically occurs during times such as fasting, illness, exercise, or sleep.
When mutations occur in the ACADVL gene, the VLCAD enzyme may not work properly or may not be produced at all. Without a functioning VLCAD enzyme, the body cannot efficiently break down certain long-chain fats to produce energy. As a result, people with VLCAD deficiency may experience energy shortages during periods when the body relies more heavily on fat for fuel. In addition, partially broken-down fatty acids can build up in the body, which may contribute to metabolic complications.
Several different types of genetic changes in the ACADVL gene can cause VLCAD deficiency. These mutations, also known as variants, affect how the VLCAD enzyme is made or how well it functions. Because many different mutations in the ACADVL gene can lead to VLCAD deficiency, the severity of symptoms can vary from person to person depending on how much enzyme activity remains.
Common types of disease-causing mutations include:
Types of ACADVL Gene Mutations
-
A single DNA change causes one amino acid in the VLCAD enzyme to be replaced with another. This can alter how the enzyme folds or functions.
-
A mutation creates a premature “stop” signal in the gene, causing the VLCAD enzyme to be shortened and usually nonfunctional.
-
A portion of the ACADVL gene is missing. This can disrupt the instructions needed to produce a working enzyme.
-
Extra pieces of DNA are added to the gene, which may disrupt the gene’s reading frame and prevent proper enzyme production.
-
These mutations affect how genetic instructions are processed before the enzyme is made, potentially leading to an incomplete or incorrect VLCAD enzyme.
How VLCAD Deficiency Is Inherited
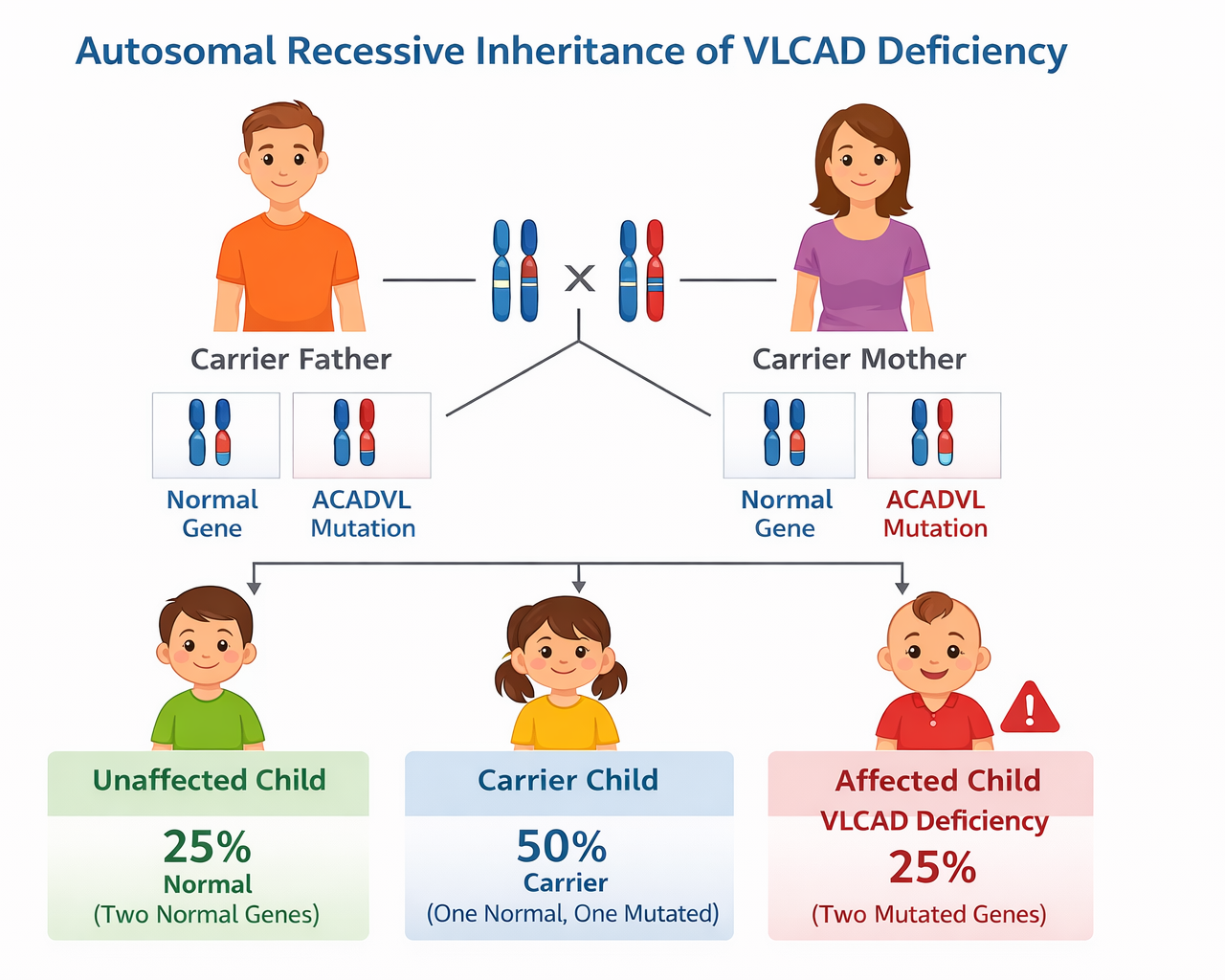
VLCAD deficiency is inherited in an autosomal recessive pattern. This means a child must inherit two altered copies of the ACADVL gene, one from each parent, to develop the condition.
Parents who carry one altered gene are called carriers. Carriers typically do not have symptoms because their other copy of the gene still produces enough enzyme for normal function.
When two carriers have a child:
25% chance the child will have VLCAD deficiency
50% chance the child will be a carrier
25% chance the child will inherit two working copies of the gene
Because carriers usually have no symptoms, many families do not know they carry the gene until a child is diagnosed.
VLCAD Genetics FAQ
-
Yes. VLCAD deficiency is inherited in an autosomal recessive pattern, meaning a child must inherit two altered copies of the ACADVL gene, one from each parent, to develop the condition.
Parents who carry one altered gene are called carriers. Carriers typically do not have symptoms because their second copy of the gene works normally.
-
VLCAD deficiency is caused by mutations in the ACADVL gene.
This gene provides instructions for making the very long-chain acyl-CoA dehydrogenase (VLCAD) enzyme, which helps break down long-chain fatty acids to produce energy in the mitochondria.
When this enzyme does not function properly, the body cannot efficiently convert certain fats into energy.
-
Yes. Most parents who carry one altered ACADVL gene do not have symptoms.
Because carriers are typically healthy, many families do not know they carry the gene until a child is diagnosed through newborn screening or metabolic testing.
-
Yes. Because carriers usually have no symptoms, many family members may carry an altered ACADVL gene without realizing it. Genetic testing can help identify carriers within a family.
-
VLCAD deficiency was first recognized as a distinct metabolic disorder in 1992, when scientists identified a previously unknown enzyme involved in long-chain fatty acid oxidation.
Shortly after, researchers identified the ACADVL gene responsible for producing the VLCAD enzyme.
-
Yes. VLCAD deficiency is included in newborn screening programs in many countries, including the United States.
Newborn screening allows doctors to detect the condition before symptoms appear, enabling early monitoring and treatment.
-
VLCAD deficiency is a genetic condition present from birth. A person is born with changes in both copies of the ACADVL gene, which affects the body’s ability to break down long-chain fatty acids for energy. However, symptoms can appear at different ages, and some individuals may not experience noticeable symptoms until later in childhood or adulthood.
-
A carrier has one normal copy of the ACADVL gene and one altered copy. Carriers typically do not have symptoms because their body can still produce enough of the VLCAD enzyme for normal energy metabolism. However, carriers can pass the altered gene to their children.
-
If both parents are carriers of an ACADVL mutation, each pregnancy has the same chance of inheritance:
25% chance the child will have VLCAD deficiency
50% chance the child will be a carrier
25% chance the child will inherit two working genes
These probabilities are the same for every pregnancy.
-
Yes. Genetic testing can identify mutations in the ACADVL gene that cause VLCAD deficiency. Testing may be performed after a positive newborn screening result, when symptoms suggest a fatty acid oxidation disorder, or when family members want to know their carrier status.
-
Yes. Different mutations in the ACADVL gene can affect how much VLCAD enzyme activity remains. Some mutations allow the enzyme to retain partial function, which may lead to milder symptoms, while others result in little or no enzyme activity and may cause more severe forms of the condition.
-
Genetic counseling can help families understand:
inheritance patterns
carrier testing
genetic test results
family planning options
Genetic counselors specialize in helping families navigate genetic conditions like VLCAD deficiency.
Because VLCAD deficiency is a rare genetic metabolic disorder, many families have questions about the ACADVL gene, inheritance patterns, and carrier status. Learning how VLCAD is passed down and how gene mutations affect energy production can help explain why the condition occurs and how it is diagnosed. Here are answers to some of the most frequently asked questions about VLCAD genetics.
Because VLCAD deficiency is inherited, families may have questions about what this diagnosis means for their child and the chances of VLCAD affecting future children. Speaking with a genetic counselor can help provide personalized guidance, explain genetic test results, and discuss options for family planning.
Types of VLCAD Deficiency
VLCAD exists on a spectrum, and not every child or individual experiences the condition in the same way. Symptoms, severity, and medical needs can vary widely.

-
This is the most serious form and is typically diagnosed in infancy, often through newborn screening or early symptoms. Children with severe VLCAD may experience:
Very low blood sugar
Heart complications (cardiomyopathy)
Feeding difficulties
Frequent hospitalizations, especially during illness
Early diagnosis and specialized metabolic care are critical for safety and stability.
-
This form is often identified in infancy or early childhood. Children may appear stable but experience symptoms during illness, fasting, or increased energy demands. Common concerns may include:
Muscle weakness
Episodes of low blood sugar
Fatigue
Hospitalizations during metabolic stress
Many children in this group require ongoing dietary management and close monitoring.
-
This form may not be diagnosed until later childhood, adolescence, or even adulthood. Individuals may experience:
Exercise intolerance
Muscle pain or muscle breakdown (rhabdomyolysis)
Fatigue with prolonged activity
Even in milder forms, illness, fasting, or extreme physical stress can still trigger serious symptoms.
One of the most challenging aspects of VLCAD deficiency is that symptoms can change over time. Growth spurts, illnesses, physical activity, and developmental stages all affect how much energy the body needs. This means a child who is stable for a period of time may still face medical risks in certain situations.
It is also important to understand that two children with VLCAD may have very different experiences. Some may have frequent hospitalizations and medical complications, while others may have fewer symptoms with proper management. Every journey is unique.